Manta Rad
The past couple of months have been particularly busy for the Manta Genetics Project. Aside from presenting at a conference for the first time (Fisheries Society of the British Isles, Plymouth), where I was fortunate enough to meet many of the people who have provided me with samples and advice over the past year, I travelled to the Royal Zoological Society of Scotland (RZSS) to visit an important partner of the genetics project. Tucked away inside RZSS Edinburgh Zoo is the WildGenes laboratory, where a small group of DNA experts are busy implementing an exciting technique known as double digest restriction-site associated DNA sequencing, or ddRADseq. This technique is an extremely important part of the genetics project, and I was at the lab to carry out some ddRAD of my own.

Concentrating hard on ddRAD library prep at RZSS. © Photo by Royal Zoological Society of Scotland.
Until relatively recently, DNA-based studies have traditionally targeted small and specific sections of the genome to analyse. These chunks of DNA are known to change between different individuals, populations or species. By sequencing them, we can find out exactly how they are changing and the effect this may be having on the species. For example, microsatellites are sections of DNA where a small sequence is repeated and the number of repeats can vary among individuals. The number of different sizes of microsatellites within a population can give us an idea of the genetic diversity of that population. However, ddRAD is a little different – and it’s exciting!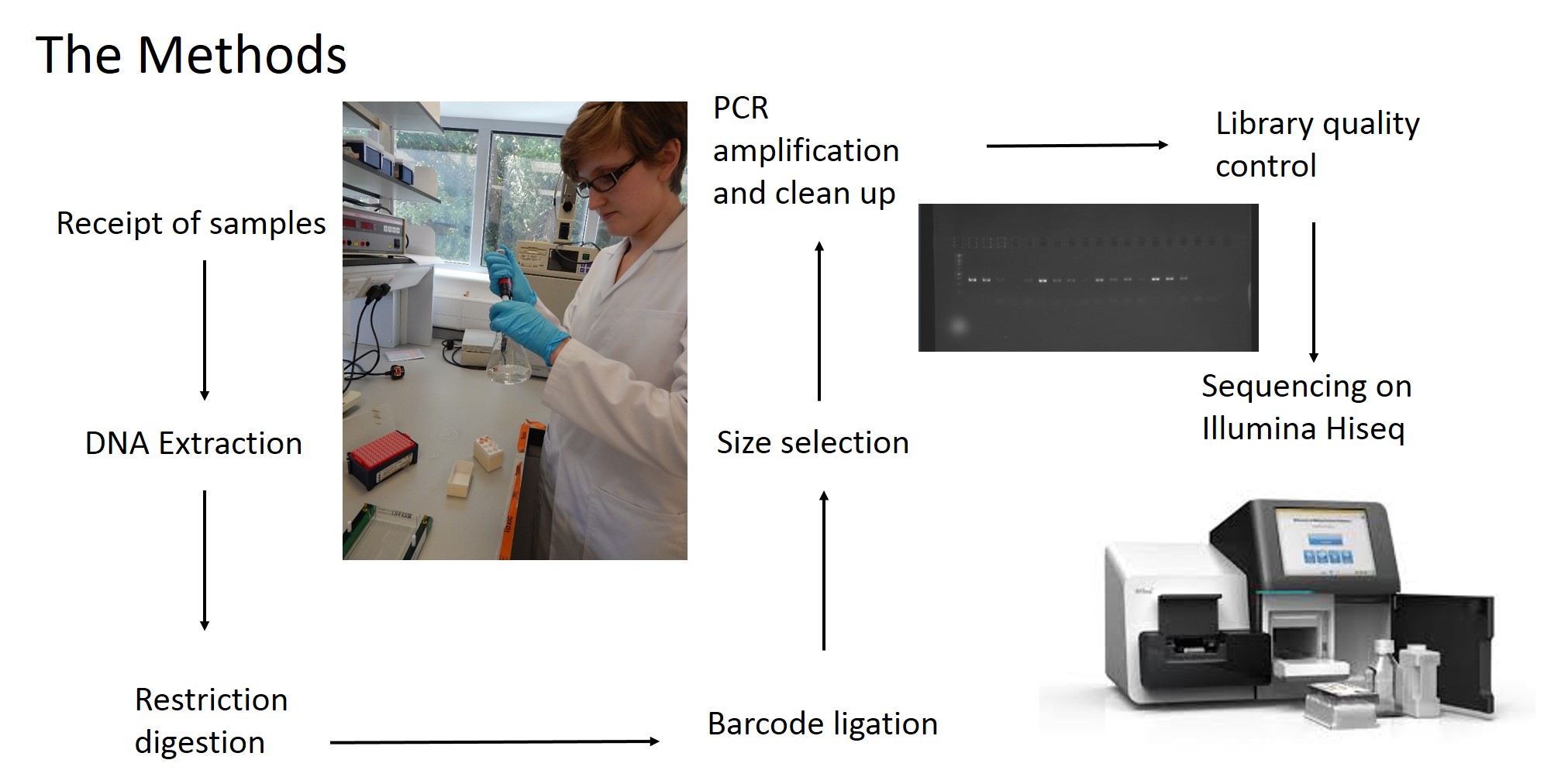
So what is ddRAD?
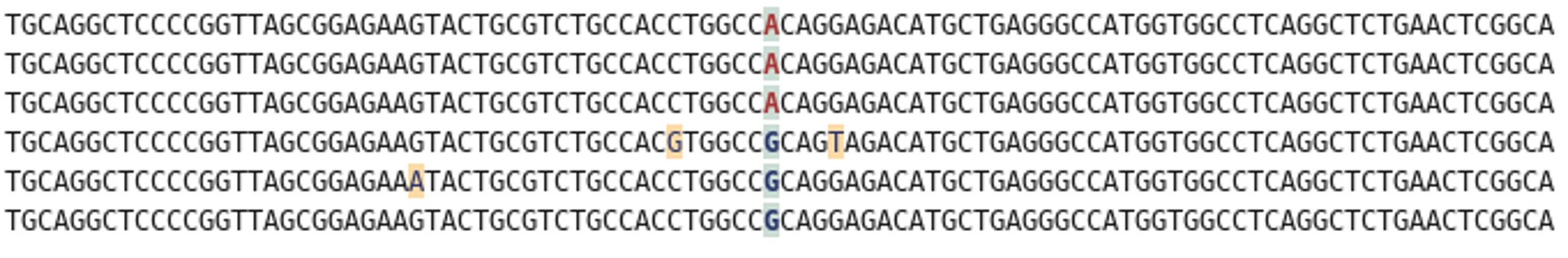
Rather than sequencing known regions of DNA like microsatellites, ddRAD takes a less targeted approach. First of all, the entire genome of an individual is cut into many small pieces. This is done using special enzymes called restriction enzymes. Each individual is then assigned a unique barcode, which is attached to the ends of each fragment via a process called ligation. Now that the barcodes are attached, and we can identify which individual a sequence comes from, we can combine the DNA from many individuals without losing track of which sequence comes from which individual. The fragments are then sorted by size, and a final set of them is chosen to make up the ‘library’, which can be sequenced on a very fancy sequencing platform (such as those developed by Illumina). What you get out of the machine are several thousand short DNA sequences that correspond to the fragments in the library. These can then be analysed using specialist software such as ‘STACKS’. STACKS matches up sequences from different individuals that are from the same place in the genome and piles them up into their own ‘stack’. Any difference in the sequence between individuals is known as a single nucleotide polymorphism (SNP) and can be identified from within the stacks. These SNPs are our genetic markers and are very useful to us.
Why is ddRAD so great?
There are many reasons why this technique is so exciting, but here are a few of my favourites:
1. You get loads of markers, and from across the whole genome.
Depending on the size of the genome of the species you’re interested in (mobula rays have quite a large genome compared to us humans), and the enzymes you choose to use, ddRAD methods have the capacity to produce sequences from about 10,000 places in the genome of a single individual. Of these, about 1,000 will have differences, or SNPs, so that’s 1,000 variable genetic markers that you’ve got information for. Let’s put that into perspective: with microsatellite-based studies, it might be possible to develop about 10 markers – 20 if you’re lucky – because that method is much more time-consuming and labour-intensive. ddRAD could give you a thousand! And that’s not even my favourite part. Different parts of the genome may be evolving more quickly or less quickly than others, affecting results where specific areas are targeted. ddRAD gets around that problem by producing markers that are distributed more or less randomly across the genome, giving us a much more rounded perspective.
2. No need for prior knowledge of the genome.
Many methods rely on the availability of a reference genome for the organism that is being studied. Sequencing entire genomes is very expensive and time-consuming (the Human Genome Project took 13 years, though it must be said that sequencing technology has advanced since then), and so reference genomes are usually only available for model organisms. ddRAD data can be obtained and analysed to yield informative results without access to a reference genome. Good news for manta genetics!
3. The same data can be used to answer different scientific questions.
Using computational methods, it is possible to look for SNPs between species, between populations, even between different colour morphs (mantas can be almost entirely black, or ‘chevron’). It’s also possible to do some clever maths to work out which SNPs are under selection – the evolutionary force that drives change. So loads of data can be generated that is useful for loads of purposes! The sequence surrounding potentially interesting SNPs can be compared to gene databases to determine whether it lies within an interesting gene.

This is all the DNA that’s needed to produce all that data!. © Photo by Jane Hosegood | Manta Trust
4. It’s cheaper than whole genome sequencing.
Even though in the past few years sequencing technologies have been developed that would blow the minds of generations of scientists before us, whole genome sequencing is still beyond the budgets of most researchers. The good news is, it’s often not needed! There are a lot of uninformative areas of the genome, so by producing a reduced representation, as we are doing with ddRADseq, resources are not wasted on sequencing large areas that don’t help answer your questions.
5. It can be done really fast!
A ddRAD library containing information about almost 100 individuals can be produced in a matter of days if you’re efficient about it. Add on some time for the sequencing of that library and you can be dealing with enormous amounts of data for the time you’ve put in.
